Data Loading Overview (All 14 Options)#
This page is the map of how Cellucid can load data.
If you are new, start here. If you already know what you want, jump to the tutorial that matches your workflow:
02_local_demo_tutorial: publish/share a dataset without running a server (GitHub-hosted exports)03_browser_file_picker_tutorial: load data from your computer using the browser file picker04_server_tutorial: run a local/remote Python server for large datasets05_jupyter_tutorial: embed Cellucid inside a notebook and interact programmatically11_custom_dataset_repository: publish and validate a public multi-dataset repository, starting from a complete reference implementation
At A Glance#
Audience
Wet lab / non-technical: choose a safe workflow and avoid common traps.
Computational: understand formats, lazy loading, and performance tradeoffs.
Power users: understand what happens under the hood so you can debug failures.
Time
Fast path decision: ~5 minutes
Full read (with edge cases): ~20–30 minutes
What you’ll learn
What “exported”, “h5ad”, and “zarr” mean in Cellucid
What “lazy loading” is and why it matters for large datasets
How to choose between File Picker, Server Mode, GitHub-hosted exports, and Jupyter
The top failure modes (and how to quickly diagnose them)
The Three Data Formats (What You Actually Load)#
Cellucid understands your data in three high-level ways:
Pre-exported folder (recommended)
Produced by
cellucid.prepare(...)in Python.Output is a directory containing binary files +
dataset_identity.json+ compact manifests.Fastest in the browser and the most stable.
Can optionally include vector fields (e.g. RNA velocity) for the overlay.
AnnData
.h5adfileA single HDF5 file.
Can be loaded in several ways:
directly in the browser (works, but has hard performance limits)
via a Python server (recommended for large datasets)
inside Jupyter (recommended for analysis workflows)
AnnData Zarr v2 store
A directory-based format made of chunked arrays.
The browser UI accepts one portable
.zarr.zip(or.zip) archive containing the complete store.Python server and Jupyter APIs accept the Zarr directory itself and materialize it eagerly with
anndata.read_zarr.
Rule of thumb: if you care about smooth interaction and reproducibility, export once using prepare().
For deeper context:
why stable IDs matter: Dataset identity (why it matters)
what files/keys are expected: Folder / file format expectations (high-level; link to spec)
when loading fails: Troubleshooting (data loading)
What “Lazy Loading” Means (In Plain Language)#
When you load a dataset, Cellucid needs several kinds of data:
Embeddings (UMAP coordinates): small-ish (n_cells × 2/3) → loaded early
Obs fields (metadata for coloring/filtering): usually manageable → loaded early
Gene expression (n_cells × n_genes): huge → ideally loaded on demand
Lazy loading means:
Cellucid loads just enough to render and interact.
When you search a gene (or request some data), it fetches only what it needs.
Why you care:
For large datasets, loading everything up front can crash the browser or take minutes.
Lazy loading keeps memory bounded and makes the UI responsive sooner.
Important nuance:
Some formats/modes are truly lazy.
Some are “lazy-ish” (metadata up front; some chunks later).
Some are not lazy in practice (browser
.h5adrequires loading the whole file).
Vector fields (velocity/drift overlay): what you need for it to appear#
Vector fields are optional per-cell vectors (e.g. RNA velocity, drift/displacement) that Cellucid can render as an animated overlay on top of your embedding.
Important rules:
Vector fields are dimension-specific: you need 2D vectors for a 2D embedding, 3D vectors for 3D, etc.
Vector fields must match the same row order as your points/cells.
The overlay only shows fields available for the current dimension.
Where vector fields come from depends on your loading format:
Exports (
prepare):dataset_identity.jsoncontains avector_fieldsblock, and binary vectors live undervectors/.AnnData (
.h5ad/.zarr/ in-memory): vector fields are discovered fromadata.obsmkeys like:velocity_umap_2d,velocity_umap_3dT_fwd_umap_2d
If you expect vector fields but don’t see the overlay toggle or dropdown:
first check your data format/naming: Folder / file format expectations (high-level; link to spec)
then check the overlay section: Vector Field / Velocity Overlay (GPU Particle Overlay)
Naming conventions:
Fast Path: Choose A Workflow (Decision Tree)#
Pick the first row that matches you.
Your situation |
Recommended workflow |
Why |
|---|---|---|
“I just want to look at my data quickly, no Python.” |
Browser File Picker |
Zero setup; good for quick preview |
“My dataset is big (hundreds of thousands to millions of cells).” |
Server Mode or pre-export + Server Mode |
True lazy loading; avoids browser memory limits |
“I’m already working in a notebook.” |
Jupyter ( |
Tight analysis loop; programmatic control |
“I want to share a dataset publicly without running a server.” |
GitHub-hosted exports |
Exact dataset-specific URL; no running server |
“I need the fastest possible web experience.” |
Pre-exported folder |
Best performance and stability |
If you’re unsure, start with Server Mode for .h5ad/.zarr, or File Picker for exported folders.
The 14 Loading Options (Complete Matrix)#
This is the canonical list used throughout the documentation.
Legend
Exported = folder created by
cellucid.prepare()Lazy genes: whether gene expression is fetched on demand (best) vs effectively “load all” (worst)
Vector fields: supported in all loading options; the overlay appears only if your dataset includes vectors for the current dimension.
# |
Where you run things |
How you point Cellucid to the data |
Data format |
Lazy genes |
Best for |
|---|---|---|---|---|---|
1 |
Cellucid web app (demo mode) |
Choose a built-in demo dataset |
Exported |
✅ |
Learning the UI with known-good data |
2 |
Cellucid web app (public GitHub) |
Connect to a public exports root (or use |
Exported |
✅ |
Sharing a dataset publicly, no running server |
3 |
Cellucid web app |
Browser Prepared picker |
Exported |
✅ |
Fast local viewing of prepared exports |
4 |
Cellucid web app |
Browser .h5ad picker |
|
❌* |
Quick preview of small |
5 |
Cellucid web app |
Browser Zarr ZIP picker |
|
✅† |
Portable Zarr viewing without Python |
6 |
Terminal CLI |
|
Exported |
✅ |
Reliable viewing of large exports |
7 |
Terminal CLI |
|
|
✅ |
Large |
8 |
Terminal CLI |
|
|
✅ |
Eager Python load with on-demand browser gene requests |
9 |
Python |
|
Exported |
✅ |
Scripting server startup |
10 |
Python |
|
|
✅ |
Scripting server startup |
11 |
Python |
|
|
✅ |
Scripting server startup |
12 |
Jupyter |
|
Exported |
✅ |
Notebook-based exploration of exports |
13 |
Jupyter |
|
|
✅ |
Notebook-based exploration of |
14 |
Jupyter |
|
|
✅ |
Notebook-based exploration of |
* Browser .h5ad loading is not truly lazy: the whole file is loaded into browser memory before use.
† Browser Zarr ZIP loading indexes and validates archive metadata before adoption, then reads gene-expression chunks on demand. Archive extraction and decoded chunk memory remain subject to the documented browser limits.
Minimal Commands (Copy/Paste)#
Install#
pip install cellucid
1) Export once (recommended)#
from cellucid import prepare
# prepare(...) writes an export folder that loads fast in the browser.
# You will typically pass:
# - obs (cell metadata)
# - var (gene metadata)
# - gene_expression (adata.X)
# - embeddings (e.g., X_umap_2d / X_umap_3d)
# - vector_fields (optional: velocity/drift overlays)
# See the prepare() documentation for full details.
2) Serve any data (auto-detected)#
cellucid serve /path/to/data.h5ad \
--dataset-name "My study" \
--dataset-id my-study-v1
cellucid serve /path/to/data.zarr \
--dataset-name "My study" \
--dataset-id my-study-v1
cellucid serve /path/to/export_dir
3) Notebook quick start#
from cellucid import show_anndata
viewer = show_anndata(
"data.h5ad",
dataset_name="My study",
dataset_id="my-study-v1",
)
# Optional: sanity-check your AnnData quickly
#
# This cell is intentionally lightweight; it's here so you can quickly
# confirm that your dataset has the minimum requirements before you pick a workflow.
from __future__ import annotations
from pathlib import Path
import numpy as np
try:
import anndata as ad
except Exception as e: # pragma: no cover
raise RuntimeError(
"anndata is required for this cell. Install with: pip install anndata"
) from e
def summarize_anndata(path: str | Path) -> dict:
path = Path(path)
adata = ad.read_h5ad(path, backed="r") # doesn't load full X into memory
try:
# Vector fields use exact dimension-suffixed obsm keys and no X_ prefix:
# <field>_umap_<dim>d (for example, velocity_umap_2d).
import re
vector_field_re = re.compile(r"^(?!X_).+_umap_[123]d$")
vector_field_obsm_keys = sorted(
[k for k in adata.obsm.keys() if vector_field_re.match(k)]
)
return {
"path": str(path),
"n_cells": int(adata.n_obs),
"n_genes": int(adata.n_vars),
"obsm_keys": list(adata.obsm.keys()),
"vector_field_obsm_keys": vector_field_obsm_keys,
"has_X": adata.X is not None,
"X_type": type(adata.X).__name__,
}
finally:
adata.file.close()
# Example usage (uncomment):
# print(summarize_anndata("/path/to/data.h5ad"))
Interface reference#
For the complete verified loading gallery, see Verified data-loading captures.
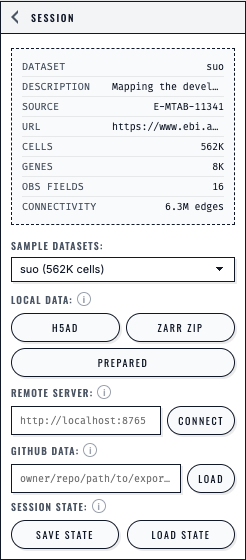
The Session panel presents each loading path separately and keeps Save State and Load State beside the dataset controls.#
Edge Cases (Read This If Something Feels “Weird”)#
Data edge cases#
No UMAP / embedding keys: your AnnData is missing
X_umap_1d,X_umap_2d, orX_umap_3dinobsm.NaN/Inf in embeddings: current readers reject the dataset before it can replace the active dataset.
Duplicate variable names: direct AnnData readers reject them because a gene request must identify exactly one column.
Huge categorical fields (e.g., 100k unique categories): legends and color assignment become unusable.
Scale edge cases#
Browser
.h5ad: if the file is large, the browser may freeze or crash due to memory pressure.Millions of cells + dense gene matrix: even with lazy loading, gene queries can be slow or memory-heavy.
Environment edge cases#
Prepared directory picker: it requires the browser’s directory-input capability. H5AD and Zarr ZIP use ordinary single-file inputs.
Corporate environments: content blockers can break
raw.githubusercontent.com(GitHub-hosted exports).
Troubleshooting (Quick Index)#
This is a compact troubleshooting index. Each follow-up notebook contains a much longer troubleshooting section.
Symptom: “Nothing loads / it spins forever”#
Likely causes
Large
.h5adopened directly in the browserCORS/network blocked (server mode or GitHub mode)
Browser GPU/WebGL context lost due to memory pressure
How to confirm
Open Developer Tools → Console → look for network errors / memory warnings
Try loading a built-in demo dataset: if demos load, your environment is probably fine
Fix
Prefer
cellucid serve data.h5ad --dataset-name "My dataset" --dataset-id my-datasetfor large.h5adPrefer exports created by
prepare()for best performance
Symptom: “I don’t see any embedding / it says no UMAP”#
Likely causes
AnnData missing
X_umap_1d,X_umap_2d, orX_umap_3dinobsm
How to confirm
In Python:
print(adata.obsm.keys())
Fix
Compute UMAP and store it in
obsmbefore viewing
Symptom: “GitHub connect says datasets.json not found”#
Likely causes
The repo path points at the wrong folder
You uploaded only a single dataset folder without a top-level
datasets.json
Fix
Create an exports root directory containing
datasets.jsonand one or more dataset subfolders.See Local & Remote Demo (Share Without Running a Server) for the exact layout and a helper function to generate
datasets.json.
Next Steps#
Choose your path:
Want to share a dataset publicly (no server)? → Local & Remote Demo (Share Without Running a Server)
Want a complete public repository to inspect and adapt? → Publish a Custom Dataset Repository
Want to load from your own machine right now? → Browser File Picker (No Python Setup)
Working with a big
.h5ad/.zarr? → Server Mode (CLI + Python)Already in notebooks? → Jupyter Integration (Notebook Embedding)